“Our lab is all about big data.”
A major cause of cancer treatment failure is the development of therapy resistance in tumors, which evolve by accumulating tumor-promoting mutations and modified chromosome structures in a subset of their cells. Overcoming such medical challenge in cancer treatment requires creative “computational analysis of big biomolecular profiles coupled with a deep understanding of cancer biology”. To contribute to the pathway to improved cancer treatments, our lab addresses basic science questions that inform on human cancers and ultimately diagnostic, prognostic and therapeutic applications in personalized medicine, through computational and statistical analysis of biomolecular data. Collaborating with the colleagues from a broad spectrum of areas (cancer biology, epigenetics, structural biology, bioimaging, tumor microenvironment, machine learning, and so on) will be essential to the successful completion of our research goal.
Research area
- Tumor evolution
- Extrachromosomal DNA
- Genomics
- Mutational processes
- Tumor microenvironment
- Biomedical data analysis system to fight diseases
- Machine learning applications in understanding diseases
- Neurodegenerative disease
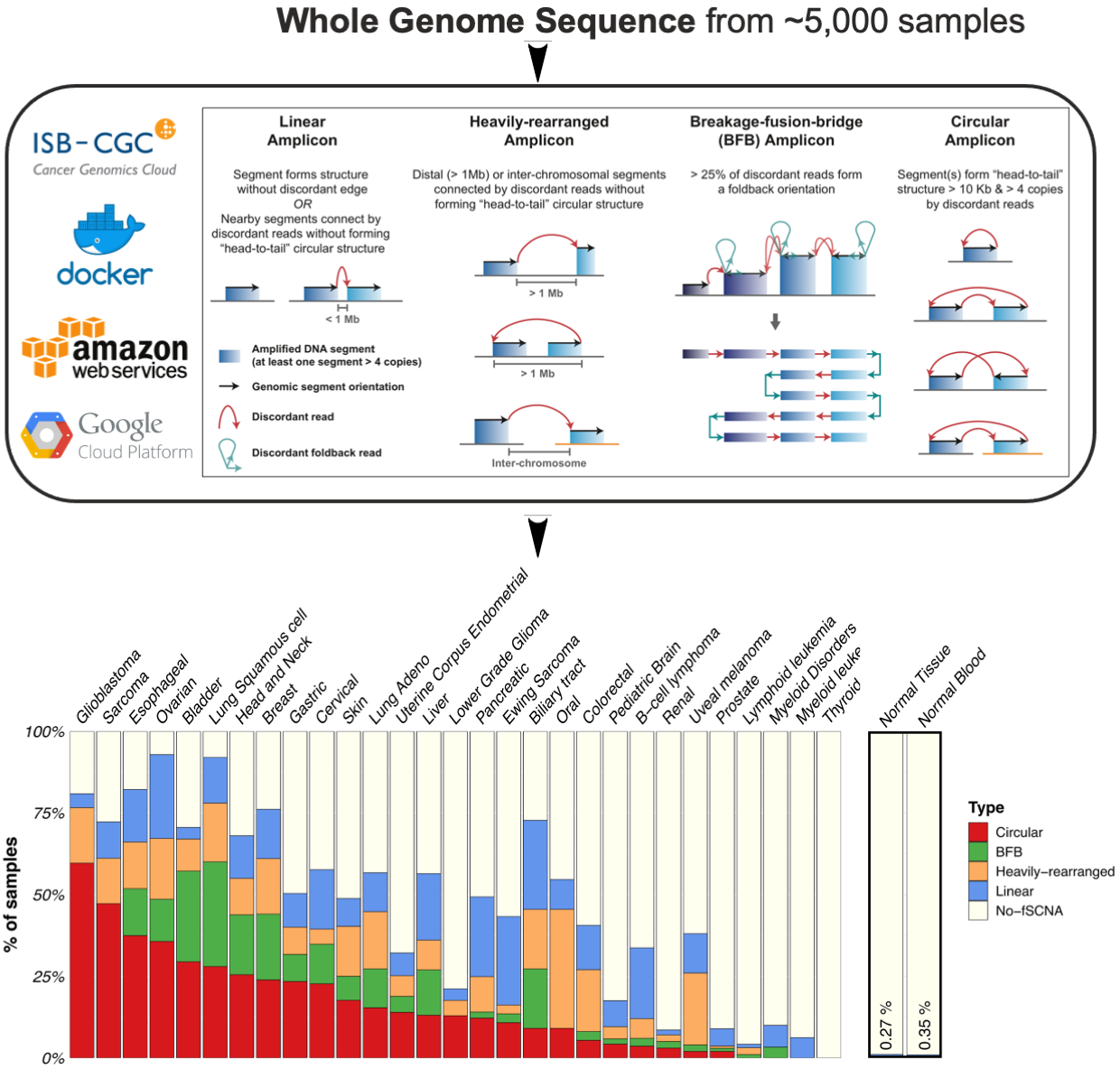
Characterization of Extrachromosomal DNA
EcDNAs in cancer were first recognized through pioneering cytopathology studies by Arthur Spriggs over 50 years ago, yet the mechanisms of ecDNA-driven tumorigenesis are still poorly studied, as are their specific roles in disease. Recently we led the first pan-cancer analysis to survey ecDNA in tumor genome sequencing data sourced from TCGA and Pan-Cancer Analysis of Whole Genomes. We were able to detect and characterize large, circular ecDNA structures from cancer patients in a total of >=5,000 samples. While it was previously thought that ecDNA was present in fewer than 1% of tumors, our study discovered ecDNAs over 25 of 29 cancer types analyzed. We also found that patients whose cancers have ecDNA have significantly shorter survival than all other cancer patients, whose tumors are driven by other molecular lesions, even when corrected for tumor type. This study provided a new window into the molecular epidemiology of ecDNA in cancer, providing a unique opportunity to study patients to better understand how and why they respond poorly to treatment. With a vast amount of sequencing data generated from diverse molecular types and advances in profiling technologies and molecular tools, it is now an excellent timing for deeply understanding ecDNAs and other forms of complex chromosomal rearrangements that can be reconciled with them in unprecedented ways.
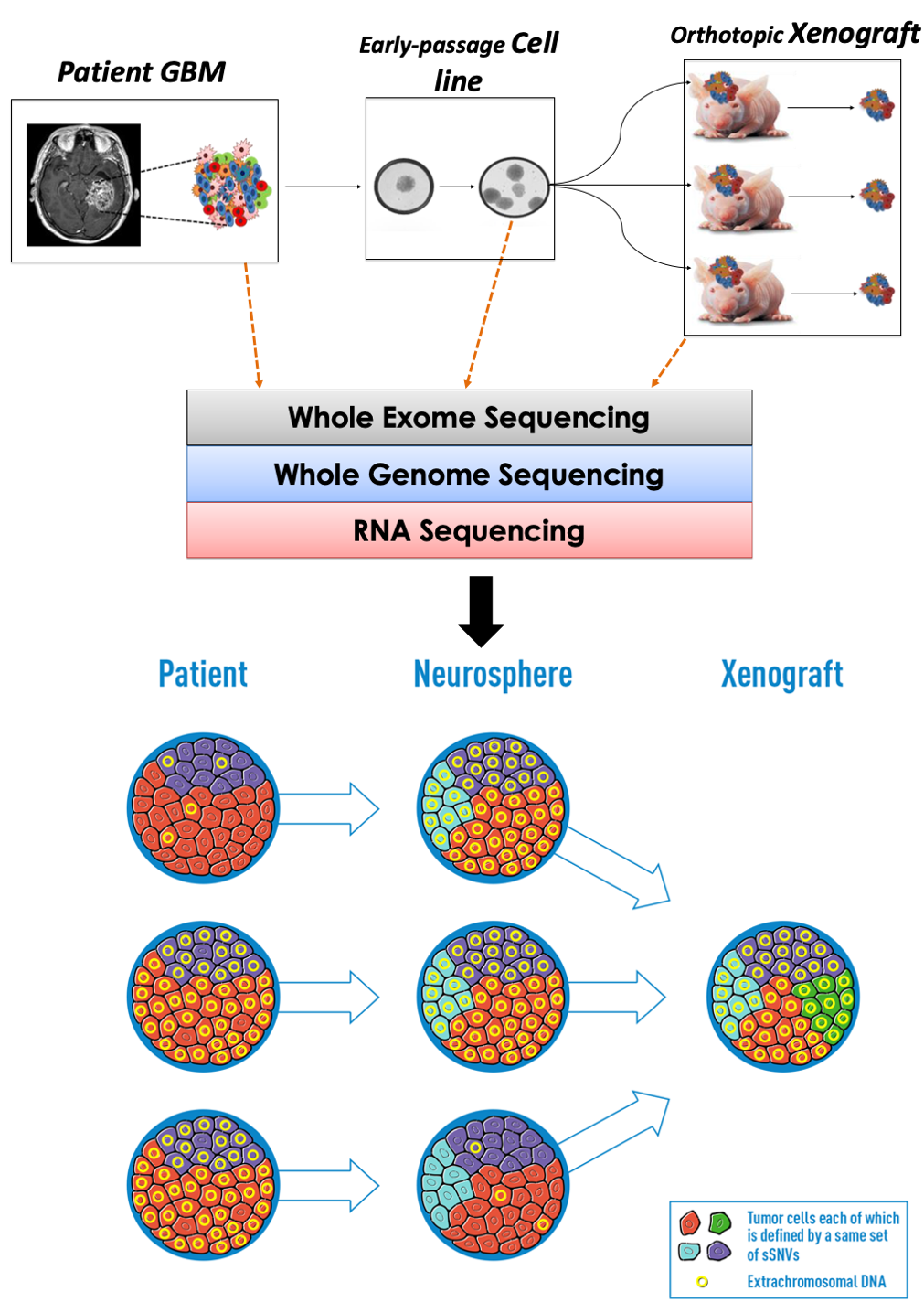
Tumor evolution and intratumoral heterogeneity
Genetic ITH and tumor evolution have been well characterized with respect to “chromosomal alterations”, but a role of “non-chromosomal alterations” in glioma evolution is not well understood. Our computational and experimental analysis of glioblastoma tumor patients and their derived model systems (neurospheres and orthotopic xenografts) identified (non-chromosomal) ecDNA amplification in these transitions, providing direct evidence that extrachromosomal amplification of oncogenes enhances intratumor heterogeneity during tumor evolution. The findings from this study change our views on the evolution of glioblastoma, having important implications for other ecDNA-carrying cancer types. Little was known about the mechanism through which these elements arise or disappear and how they become fixed across a cancer cell population. Our analysis provides a comprehensive study of the fate of chromosomal alterations and ecDNA oncogene amplifications in glioblastoma in a panel of tumors and derivative models, and the work moving forward will help explain why these cancers are difficult to treat and evolve therapy resistance so rapidly.
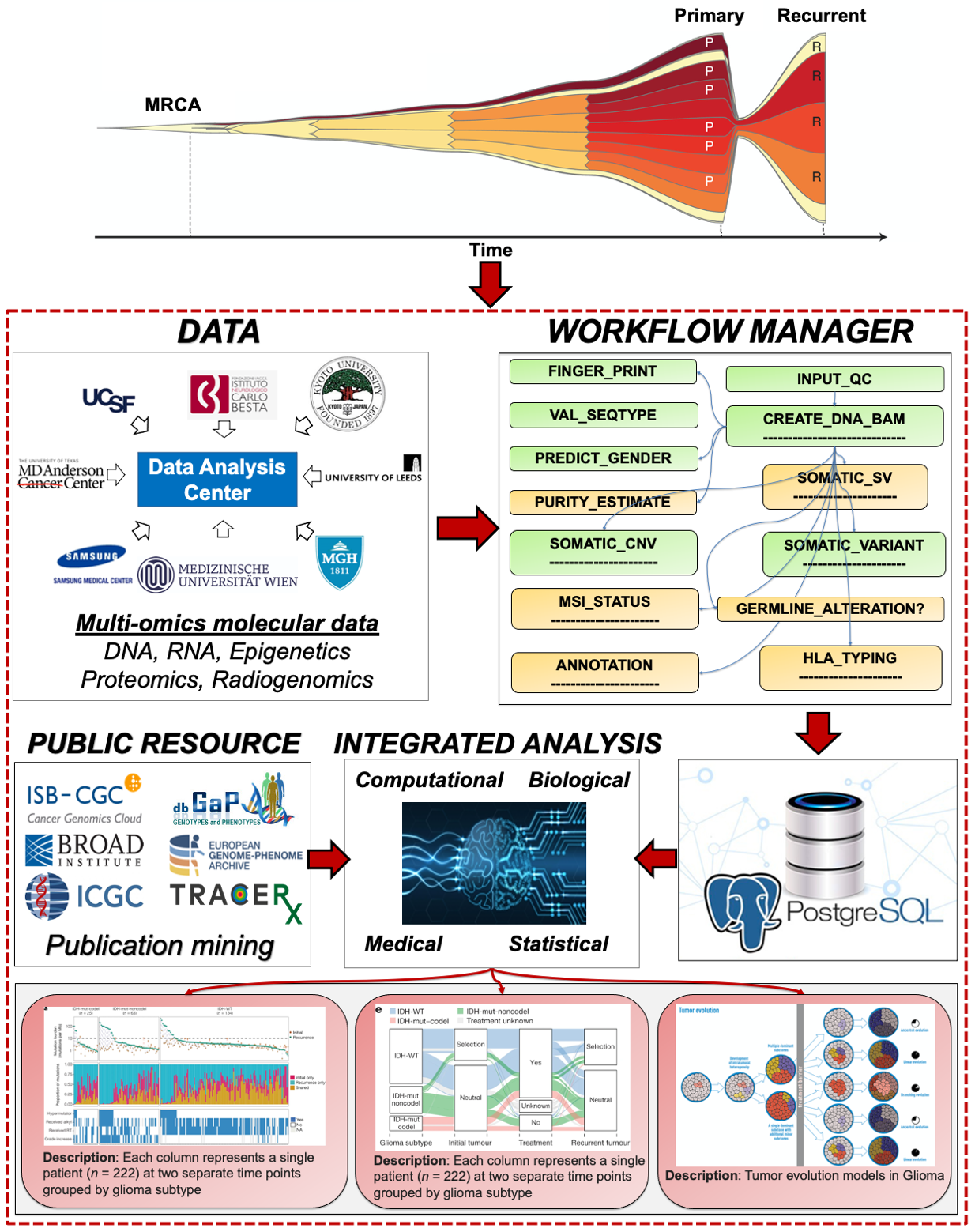
Biomedical data analysis system to fight diseases
Previously, I was able to be involved in the genomic characterization of several tumor types from The Cancer Genome Atlas (TCGA). This unique experience laid the foundation for the Glioma Longitudinal AnalySiS (GLASS) consortium, a multi-institutional effort composed of more than 34 academic hospitals, universities, and research institutes from 12 countries, aiming to comprehensively characterize the evolutionary profiles from analysis of primary and matched recurrent glioma tumor samples from a large cohort in each of the three major glioma molecular subtypes that were recently updated in the World Health Organization (WHO) classification. I am a founding member of the consortium in the data analysis committee.
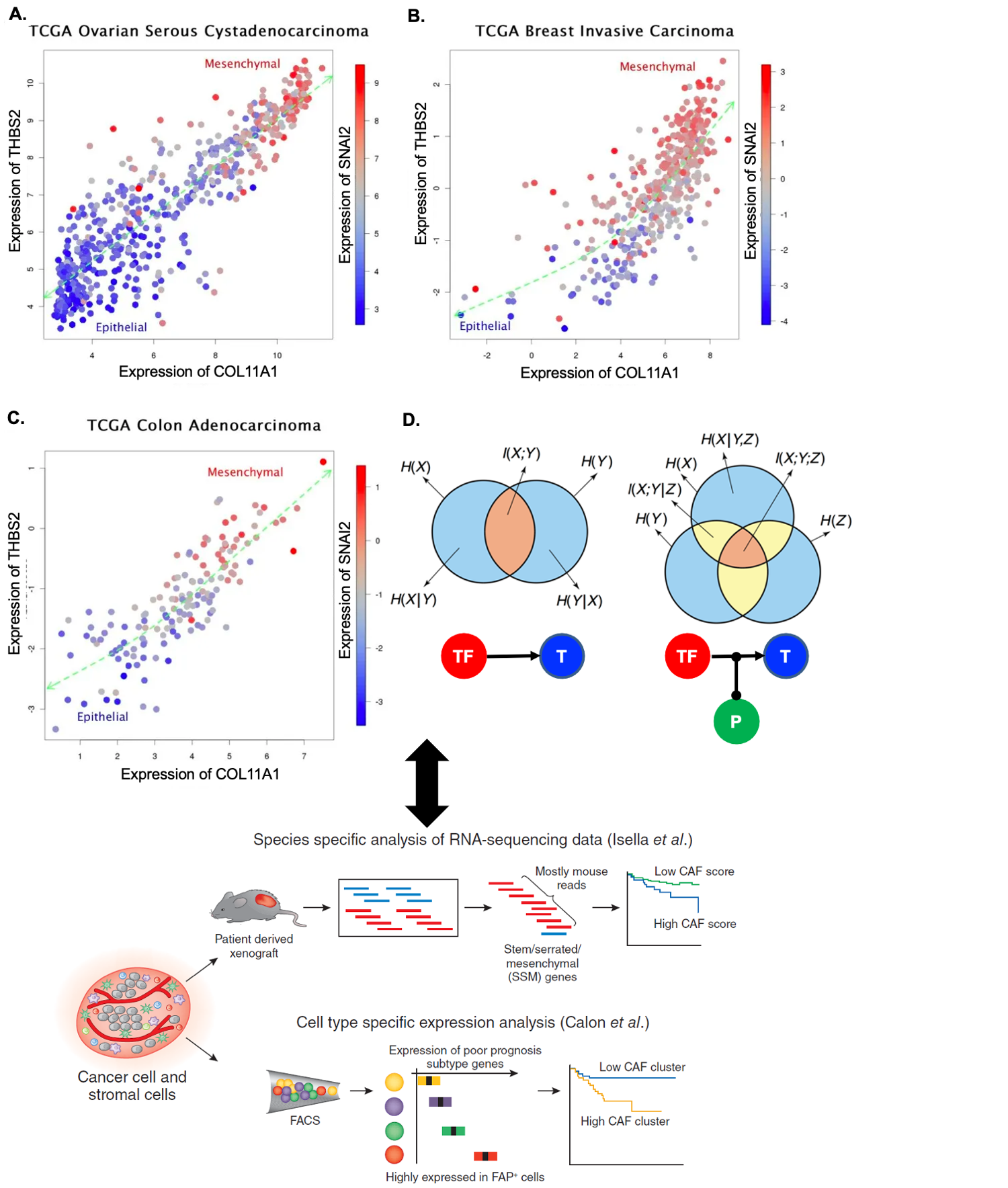
Machine learning applications in understanding diseases
I discovered the first pan-cancer invasion-associated gene signature that was coordinately over-expressed in a subset of patients from different cancer types, but only when a particular stage specific to each tumor type was exceeded. This pan-cancer signature represents a key “bioinformatic hallmark of cancer invasion”, and it was used in a nearly identical form as one of the core signatures in the best-performing computational model for breast cancer prognosis in the Sage Bionetworks–DREAM Breast Cancer Prognosis Challenge. Since my previous discovery, many independent research results (mostly from analyzing bulk microarray data profiled from xenografts with fibroblast-specific markers) have been referring to a particular type of cancer-associated fibroblasts associated with invasiveness, metastasis and resistance to therapy, present in nearly identical form over many individual types of solid cancer (i.e. breast, ovarian, colon, pancreatic cancers). This multi-cancer signature also served as biomarkers in individual cancer types in some studies, highly associated with clinical outcomes; however, their cellular origin, and precise underlying mechanisms are elusive. We will tackle the ten-year-old question, which will provide additional testable hypotheses for better understanding interactions with cancer cells and infiltrating cells and developing invasiveness-inhibiting therapeutics applicable to multiple cancer types.